What is HiCStuff?
HiCstuff is a Hi-C analysis python library which generates contact matrices from input Hi-C fastq files & integrates modules to manipulate them.
This web server is based on the
hicstuff pipeline module, providing the (often most resource-consuming) first steps in Hi-C data analysis, namely
mapping of reads onto a reference genome, filtering out of spurious 3C events, binning of
matrices based on restriction enzymes used and plotting of the resulting contact matrix.
General outline
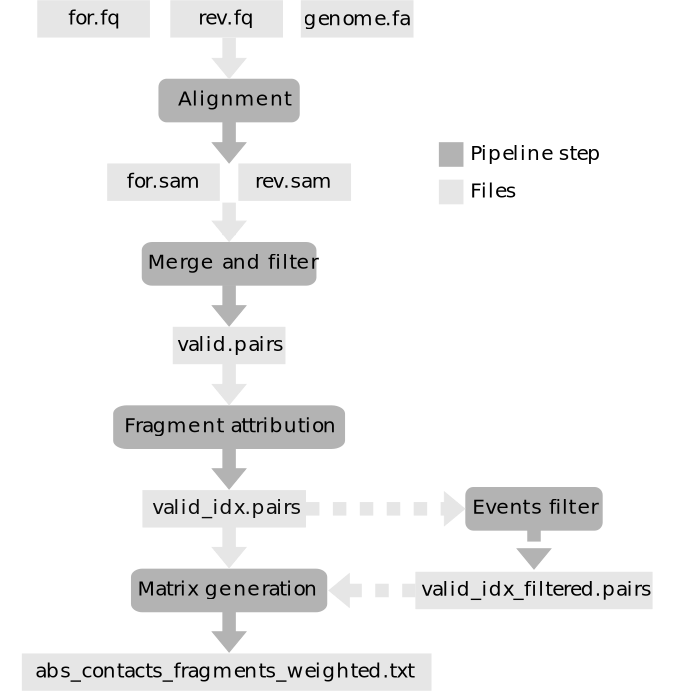
What next?
Although the first steps in Hi-C data analysis are quite general, the analysis that you will perform as a second step on the generated contact matrices are highly dependent on the experimental approach and the biological question you want to answer. We suggest you follow the progressive OHCA tutorial to continue your analysis using the programming language R.
If you have trouble installing the right packages on a local R session, you can try going through the IFB's Open OnDemand facility (it's free for academics, you just have to create an account first). Once logged in, you can open an RStudio Server session and run:
library(HiCExperiment) library(HiCool) library(HiContacts)
https://ondemand.cluster.france-bioinformatique.fr/
To import the generated matrix in an R session, download the zip archive on your results page and decompress it, then open an R session and type:
cool_file <- CoolFile("/path/to/your/file.mcool")
hic <- import(cool_file)NB: Make sure to load the required libraries first (of which, "HiCExperiment"), cf. corresponding tutorial page.
Tools & versions
Tools used for the analysis pipeline
- HiCstuff (v3.2.1):
Cyril Matthey-Doret, Lyam Baudry, Amaury Bignaud, et al.
hicstuff: Simple library/pipeline to generate and handle Hi-C data.
(2020).
DOI: http://doi.org/10.5281/zenodo.4066363 -
Chromosight (1.6.3):
Matthey-Doret, C., Baudry, L., Breuer, A. et al.
Computer vision for pattern detection in chromosome contact maps.
Nat Commun 11, 5795 (2020).
DOI: https://doi.org/10.1038/s41467-020-19562-7 -
OHCA (packages, tutorial and user manual):
Serizay J, Matthey-Doret C, Bignaud A, Baudry L, Koszul R.
Orchestrating chromosome conformation capture analysis with Bioconductor.
Nature Communications, 15, 1-9 (2024).
DOI: https://doi.org/10.1038/s41467-024-44761-x -
Samtools (1.11):
Petr Danecek, James K Bonfield, Jennifer Liddle, John Marshall, et al.
Twelve years of SAMtools and BCFtools
GigaScience, Volume 10, Issue 2, February 2021, giab008
DOI: https://doi.org/10.1093/gigascience/giab008 -
Bedtools:
Aaron R. Quinlan, Ira M. Hall,
BEDTools: a flexible suite of utilities for comparing genomic features,
Bioinformatics, Volume 26, Issue 6, March 2010, Pages 841–842
DOI: https://doi.org/10.1093/bioinformatics/btq033 -
BigWig & BigBed tools:
Kent WJ, Zweig AS, Barber G, Hinrichs AS, Karolchik D.
BigWig and BigBed: enabling browsing of large distributed data sets.
Bioinformatics. 2010 Sep 1;26(17):2204-7.
DOI: https://doi.org/10.1093/bioinformatics/btq351 -
3DGB:
Poinsignon T., Gallopin M., Grognet P., Malagnac F., Lelandais G., & Poulain P.
3D models of fungal chromosomes to enhance visual integration of omics data.
NAR Genomics and Bioinformatics. 2023 5(4).
DOI: https://doi.org/10.1093/nargab/lqad104 -
PASTIS:
Varoquaux N., Ay F., Noble W. S., & Vert J.-P.
A statistical approach for inferring the 3D structure of the genome.
Bioinformatics. 2014 30(12), i26–i33.
DOI: https://doi.org/10.1093/bioinformatics/btu268
Tools used for the web pages
This site was generated using Django and django modules.
-
Django v4.1.6 [Computer Software]. (2023).
https://www.djangoproject.com/ -
Bootstrap v4.1.3
Github. https://github.com/twbs/bootstrap -
jQuery v3.7.1
Github. https://github.com/jquery/jquery -
NGL Viewer
DOI: https://doi.org/10.1093/nar/gkv402
Contact information
Acknowledgements
We thank Amaury Bignaud, and Martial Marbouty and his team at the Institut Pasteur for his help in setting up the analysis pipeline and for sharing his scripts. We also thank Fabrice Confalonieri and Gaëlle Lelandais from the I2BC for his input on setting up this service.