Getting started with Snakemake
For this practical exercise, we will:
- access snakemake (setup)
- create a first snakefile with a single rule (o1)
- upscale your pipeline with a second input (o2)
- add a second rule to create a first workflow (o3)
- discover & understand the use of a target rule (o4)
- understand how rules are linked (o4)
- learn how to generalise inputs and outputs of rules (#wildcards) (o5 & o6)
- get accustomed to using wildcards within a snakemake rule (o5 & o6)
- learn how to visualise your pipeline (recap)
- learn how to simulate the execution with dry-run (recap)
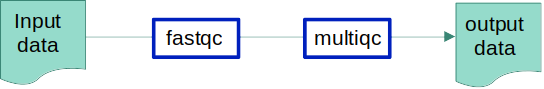
Our input: bulk RNA-seq data in fastq format
Our objective: to evaluate the quality of our data
Our tools: FastQC and MultiQC are tools commonly used to analyse the quality of sequencing data, we would like to run these within a Snakemake pipeline
Our final objective is to create a snakefile to manage this small workflow:
How this exercise is organised:
We will be building the pipeline progressively together. Each step will reply to an objective. Thus, we will be doing several cycles of executing snakemake, observing the results and improving the code. Each code version will be noted ex1_oX.smk, with X a progressive digit corresponding to the objective number.