Getting started with Snakemake
Workflows
A workflow (or pipeline) is a sequence of operations used to complete a process. In biology, we frequently use high-throughput technology, generating a large amount of data, and use a sizable amount of different bioinformatics tools, often specialised in a single task, to analyse the resulting data. This can lead to the creation of complex analysis workflows that are difficult to manage manually.
Below is an example of a fairly simple workflow in which “n” tools are sequentially applied to the initial input data. Here, the output of the first tool becomes the input for the next one, etc. until we get the desired “output data”. This process will also generate a certain number of intermediate or temporary files (“tmp data”) that might or might not be interesting to keep.
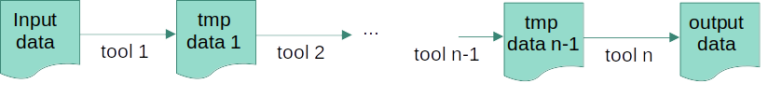
Workflow managers
Why? Workflow management tools, such as Snakemake, are there to minimise the number of manual steps that are required to execute an analysis workflow. They simplify pipeline development, maintenance, and use, by dealing with task parallelisation, resuming of failed runs or steps and tracking of parameters for example, and by simply making your code generally less complex to read and modular. They also make your pipeline more easily scalable to large sets of data.
What one to use? Many workflow management tools exist (https://github.com/pditommaso/awesome-pipeline), some being more specific to the bioinformatics community and often compatible with FAIR-applying habits to make the workflow more portable and reproducible, such as the use of containers (e.g. apptainer/singularity, docker) or package managers (e.g. conda). Which workflow management tool to use will depend on your application and the properties you’re interested in (e.g. ease of use, portability, scalability, availability, etc.), see Table 1 in Wratten et al., 2021.
Popular examples: The most commonly used workflow managers in the bioinformatics field at the moment are graphical and DSL (Domain-specific language) workflow managers. Graphical managers, of which a popular example is Galaxy, don’t require programming experience (i.e. “point-and-click”). On the other hand, DSL-based workflow managers are designed for bioinformaticians because they require a minimum of programming experience. Nextflow and Snakemake are very popular examples of such managers, having the largest community and many ready-to-use pipelines available.
About Snakemake
This course will focus on Snakemake. It’s maintained by an academic lab and is open-source. It’s also one of the most popular DSL-based workflow management tools and has a large community providing an abundance of learning resources and direct access to community support.
Snakemake’s language is similar to that of standard Python syntax and works backwards by requesting output files and defining each step required to produce them (similar to Make). Thus, in the example diagramme above, it will start by checking if “output data” exists, and if not, it will check if “tmp data n-1” exists to generate “output data”, and so on.
Existing tools and pipelines written in other scripting languages can be easily incorporated into Snakemake pipelines. Snakemake also enables between-workflow caching to avoid recomputation of shared steps between pipelines.
Snakemake vs Nextflow
There is no clear winner. Snakemake and Nextflow are both open-source, they were created roughly at the same time and offer similar solutions to workflow management. They have both thrived since then with a large community of users on either side.
If you have to decide between one of the two, try to pick the same one as your colleagues or close community so that you can more easily share your workflows and get help.
Some of the differences between Nextflow and Snakemake include:
- Language: Snakemake is based on Python, which might be more familiar to some, whereas Nextflow is based on Groovy, which might make it more difficult and longer to master, depending on your background.
- Approach: Nextflow uses a “top-down” approach respecting the natural flow of data analysis, whereas Snakemake uses a “bottom-up” approach, which implies the pre-estimation of all dependencies, starting from the expected results and working backwards to the raw data. This, apparently, could be a limiting factor for very big calculations and workflows (only for extreme cases).
- Output definition: In Snakemake, users have to explicitly define output file names and folders (which, even if it can be tedious, can make a pipeline easier to understand for some). This is because it relies on these files to determine when to run each step of the pipeline. Having a “top-down” approach, Nextflow doesn’t need outputs to be explicitly named.
- Dry runs: Dry runs are useful to test workflows, in Snakemake, this can be done without any data, whereas in Nextflow, you have to provide at least a small dataset (but this is probably a good idea anyway to test for further bugs).
- Catalog of published & curated pipelines: Nextflow’s user community project (nf-core) is more largely known, but an equivalent and less known catalog also exists for Snakemake (Snakemake workflows).
The above statements were largely inspired by the following reference:
Wratten, L., Wilm, A. & Göke, J. Reproducible, scalable, and shareable analysis pipelines with bioinformatics workflow managers. Nat Methods 18, 1161–1168 (2021). doi: 10.1038/s41592-021-01254-9
Other references include the following blog posts: