Getting started with Snakemake
Objective 3
ex1_o3.smk in which we add a new rule which will run MultiQC on a list of all output files of FastQC.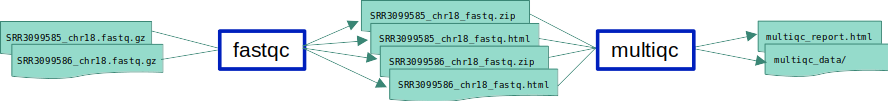
Where to start?
MultiQC is a tool used to aggregate the results of multiple other tools into a single html file.
- Input files: FastQC’s zip file outputs.
- MultiQC command:
multiqc *fastqc.zip - Expected output: 2 files:
multiqc_report.htmland amultiqc_datarepository
NB: when the output is a directory, you have to specify this using thedirectory()function. In this case, you would have to put:directory("multiqc_data")
Your code for ex1_o3.smk should look like this:
rule fastqc:
input:
"Data/SRR3099585_chr18.fastq.gz",
"Data/SRR3099586_chr18.fastq.gz",
output:
"FastQC/SRR3099585_chr18_fastqc.zip",
"FastQC/SRR3099585_chr18_fastqc.html",
"FastQC/SRR3099586_chr18_fastqc.zip",
"FastQC/SRR3099586_chr18_fastqc.html",
shell: "fastqc --outdir FastQC {input}"
rule multiqc:
input:
"FastQC/SRR3099585_chr18_fastqc.zip",
"FastQC/SRR3099586_chr18_fastqc.zip",
output:
"multiqc_report.html",
directory("multiqc_data")
shell:
"multiqc {input}"Test the script
Next, let’s check again if your pipeline works:
snakemake -s ex1_o3.smk --cores 1 -p
You should see something similar to the following output on your screen:
Assuming unrestricted shared filesystem usage for local execution.
Building DAG of jobs...
Using shell: /usr/bin/bash
Provided cores: 1 (use --cores to define parallelism)
Rules claiming more threads will be scaled down.
Job stats:
job count
------ -------
fastqc 1
total 1
Select jobs to execute...
Execute 1 jobs...
[Tue Feb 20 14:07:33 2024]
localrule fastqc:
input: Data/SRR3099585_chr18.fastq.gz, Data/SRR3099586_chr18.fastq.gz
output: FastQC/SRR3099585_chr18_fastqc.zip, FastQC/SRR3099585_chr18_fastqc.html, FastQC/SRR3099586_chr18_fastqc.zip, FastQC/SRR3099586_chr18_fastqc.html
jobid: 0
reason: Code has changed since last execution
resources: tmpdir=/var/tmp/pbs.743371.pbsserver
fastqc --outdir FastQC Data/SRR3099585_chr18.fastq.gz Data/SRR3099586_chr18.fastq.gz
Started analysis of SRR3099585_chr18.fastq.gz
Approx 5% complete for SRR3099585_chr18.fastq.gz
Approx 10% complete for SRR3099585_chr18.fastq.gz
Approx 15% complete for SRR3099585_chr18.fastq.gz
Approx 20% complete for SRR3099585_chr18.fastq.gz
Approx 25% complete for SRR3099585_chr18.fastq.gz
Approx 30% complete for SRR3099585_chr18.fastq.gz
Approx 35% complete for SRR3099585_chr18.fastq.gz
Approx 40% complete for SRR3099585_chr18.fastq.gz
Approx 45% complete for SRR3099585_chr18.fastq.gz
Approx 50% complete for SRR3099585_chr18.fastq.gz
Approx 55% complete for SRR3099585_chr18.fastq.gz
Approx 60% complete for SRR3099585_chr18.fastq.gz
Approx 65% complete for SRR3099585_chr18.fastq.gz
Approx 70% complete for SRR3099585_chr18.fastq.gz
Approx 75% complete for SRR3099585_chr18.fastq.gz
Approx 80% complete for SRR3099585_chr18.fastq.gz
Approx 85% complete for SRR3099585_chr18.fastq.gz
Approx 90% complete for SRR3099585_chr18.fastq.gz
Approx 95% complete for SRR3099585_chr18.fastq.gz
Analysis complete for SRR3099585_chr18.fastq.gz
Started analysis of SRR3099586_chr18.fastq.gz
Approx 5% complete for SRR3099586_chr18.fastq.gz
Approx 10% complete for SRR3099586_chr18.fastq.gz
Approx 15% complete for SRR3099586_chr18.fastq.gz
Approx 20% complete for SRR3099586_chr18.fastq.gz
Approx 25% complete for SRR3099586_chr18.fastq.gz
Approx 30% complete for SRR3099586_chr18.fastq.gz
Approx 35% complete for SRR3099586_chr18.fastq.gz
Approx 40% complete for SRR3099586_chr18.fastq.gz
Approx 45% complete for SRR3099586_chr18.fastq.gz
Approx 50% complete for SRR3099586_chr18.fastq.gz
Approx 55% complete for SRR3099586_chr18.fastq.gz
Approx 60% complete for SRR3099586_chr18.fastq.gz
Approx 65% complete for SRR3099586_chr18.fastq.gz
Approx 70% complete for SRR3099586_chr18.fastq.gz
Approx 75% complete for SRR3099586_chr18.fastq.gz
Approx 80% complete for SRR3099586_chr18.fastq.gz
Approx 85% complete for SRR3099586_chr18.fastq.gz
Approx 90% complete for SRR3099586_chr18.fastq.gz
Approx 95% complete for SRR3099586_chr18.fastq.gz
Analysis complete for SRR3099586_chr18.fastq.gz
[Tue Feb 20 14:07:45 2024]
Finished job 0.
1 of 1 steps (100%) done
Complete log: .snakemake/log/2024-02-20T140733.574316.snakemake.log
Observe the output
Wait, what??! What about my new multiqc rule?
Expected behaviour:
Job stats:
job count
------ -------
fastqc 1
multiqc 1
total 1
Current behaviour:
Job stats:
job count
------ -------
fastqc 1
total 1
By default, Snakemake will only execute the first rule it encounters in your Snakefile, it’s called the target rule. If (and only if) the necessary input files to execute this rule are missing, will it scan the other rules in your Snakefile to generate them. In our case, the fastqc rule is the target rule as it’s written first. Since all the necessary input files are already available for the fastqc rule, Snakemake doesn’t execute any of the other rules in the file. Let’s see in the next objective how to fix this.