Getting started with Snakemake
Objective 5
ex1_o5.smk in which we add an extra input to the fastqc rule.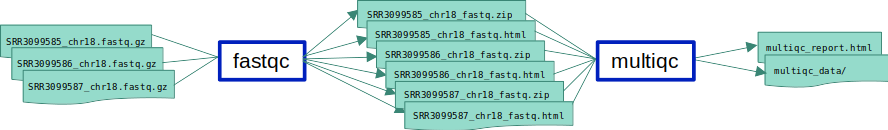
Where to start?
expand() function to manage all your input RNA-seq files at once.So, how should we go about it?
- Create a list: create a Python list at the beginning of the snakefile containing all the base names of your input files (don’t include the
.fastq.gzsuffix)
NB: in Python, a list of strings can be defined like this:list_name = ["item1","item2",...,"itemN"] - Integrate expand(): replace the list of file names in the input and output directives of your rules by the
expand()function.
For example, the input directive of your fastqc rule could look like this:
expand("Data/{sample}",sample=list_name)
Explanation: This function expects a string as input defining the file paths, in which you replace the variable parts by “placeholders” called wildcards. In this case, there is only one, that we named sample and that should be written between braces: {sample}.
As further inputs to this function, we also have to specify by what elements we would like to replace each wildcard with. In this case, we have to give the function our list of base names (sample=list_name)
Your code for ex1_o5.smk should look like this (we added the SRR3099587_chr18 sample to the previous 2):
SAMPLES=["SRR3099585_chr18","SRR3099586_chr18","SRR3099587_chr18"] rule all: input: expand("FastQC/{sample}_fastqc.html", sample=SAMPLES), expand("FastQC/{sample}_fastqc.zip", sample=SAMPLES), "multiqc_report.html",
"multiqc_data", rule fastqc: input: expand("Data/{sample}.fastq.gz", sample=SAMPLES) output: expand("FastQC/{sample}_fastqc.zip", sample=SAMPLES), expand("FastQC/{sample}_fastqc.html", sample=SAMPLES) shell: "fastqc --outdir FastQC {input}" rule multiqc: input: expand("FastQC/{sample}_fastqc.zip", sample = SAMPLES) output: "multiqc_report.html", directory("multiqc_data") shell: "multiqc {input}"
Test the script
Next, let’s run your pipeline again:
snakemake -s ex1_o5.smk --cores 1 -p
You should see something similar to the following output on your screen:
Assuming unrestricted shared filesystem usage for local execution.
Building DAG of jobs...
Using shell: /usr/bin/bash
Provided cores: 1 (use --cores to define parallelism)
Rules claiming more threads will be scaled down.
Job stats:
job count
------- -------
all 1
fastqc 1
multiqc 1
total 3
Select jobs to execute...
Execute 1 jobs...
[Tue Feb 20 14:22:40 2024]
localrule fastqc:
input: Data/SRR3099585_chr18.fastq.gz, Data/SRR3099586_chr18.fastq.gz, Data/SRR3099587_chr18.fastq.gz
output: FastQC/SRR3099585_chr18_fastqc.zip, FastQC/SRR3099586_chr18_fastqc.zip, FastQC/SRR3099587_chr18_fastqc.zip, FastQC/SRR3099585_chr18_fastqc.html, FastQC/SRR3099586_chr18_fastqc.html, FastQC/SRR3
099587_chr18_fastqc.html
jobid: 1
reason: Missing output files: FastQC/SRR3099587_chr18_fastqc.html, FastQC/SRR3099587_chr18_fastqc.zip; Set of input files has changed since last execution
resources: tmpdir=/var/tmp/pbs.743371.pbsserver
fastqc --outdir FastQC Data/SRR3099585_chr18.fastq.gz Data/SRR3099586_chr18.fastq.gz Data/SRR3099587_chr18.fastq.gz
Started analysis of SRR3099585_chr18.fastq.gz
Approx 5% complete for SRR3099585_chr18.fastq.gz
Approx 10% complete for SRR3099585_chr18.fastq.gz
Approx 15% complete for SRR3099585_chr18.fastq.gz
Approx 20% complete for SRR3099585_chr18.fastq.gz
Approx 25% complete for SRR3099585_chr18.fastq.gz
Approx 30% complete for SRR3099585_chr18.fastq.gz
Approx 35% complete for SRR3099585_chr18.fastq.gz
Approx 40% complete for SRR3099585_chr18.fastq.gz
Approx 45% complete for SRR3099585_chr18.fastq.gz
Approx 50% complete for SRR3099585_chr18.fastq.gz
Approx 55% complete for SRR3099585_chr18.fastq.gz
Approx 60% complete for SRR3099585_chr18.fastq.gz
Approx 65% complete for SRR3099585_chr18.fastq.gz
Approx 70% complete for SRR3099585_chr18.fastq.gz
Approx 75% complete for SRR3099585_chr18.fastq.gz
Approx 80% complete for SRR3099585_chr18.fastq.gz
Approx 85% complete for SRR3099585_chr18.fastq.gz
Approx 90% complete for SRR3099585_chr18.fastq.gz
Approx 95% complete for SRR3099585_chr18.fastq.gz
Analysis complete for SRR3099585_chr18.fastq.gz
Started analysis of SRR3099586_chr18.fastq.gz
Approx 5% complete for SRR3099586_chr18.fastq.gz
Approx 10% complete for SRR3099586_chr18.fastq.gz
Approx 15% complete for SRR3099586_chr18.fastq.gz
Approx 20% complete for SRR3099586_chr18.fastq.gz
Approx 25% complete for SRR3099586_chr18.fastq.gz
Approx 30% complete for SRR3099586_chr18.fastq.gz
Approx 35% complete for SRR3099586_chr18.fastq.gz
Approx 40% complete for SRR3099586_chr18.fastq.gz
Approx 45% complete for SRR3099586_chr18.fastq.gz
Approx 50% complete for SRR3099586_chr18.fastq.gz
Approx 55% complete for SRR3099586_chr18.fastq.gz
Approx 60% complete for SRR3099586_chr18.fastq.gz
Approx 65% complete for SRR3099586_chr18.fastq.gz
Approx 70% complete for SRR3099586_chr18.fastq.gz
Approx 75% complete for SRR3099586_chr18.fastq.gz
Approx 80% complete for SRR3099586_chr18.fastq.gz
Approx 85% complete for SRR3099586_chr18.fastq.gz
Approx 90% complete for SRR3099586_chr18.fastq.gz
Approx 95% complete for SRR3099586_chr18.fastq.gz
Analysis complete for SRR3099586_chr18.fastq.gz
Started analysis of SRR3099587_chr18.fastq.gz
Approx 5% complete for SRR3099587_chr18.fastq.gz
Approx 10% complete for SRR3099587_chr18.fastq.gz
Approx 15% complete for SRR3099587_chr18.fastq.gz
Approx 20% complete for SRR3099587_chr18.fastq.gz
Approx 25% complete for SRR3099587_chr18.fastq.gz
Approx 30% complete for SRR3099587_chr18.fastq.gz
Approx 35% complete for SRR3099587_chr18.fastq.gz
Approx 40% complete for SRR3099587_chr18.fastq.gz
Approx 45% complete for SRR3099587_chr18.fastq.gz
Approx 50% complete for SRR3099587_chr18.fastq.gz
Approx 55% complete for SRR3099587_chr18.fastq.gz
Approx 60% complete for SRR3099587_chr18.fastq.gz
Approx 65% complete for SRR3099587_chr18.fastq.gz
Approx 70% complete for SRR3099587_chr18.fastq.gz
Approx 75% complete for SRR3099587_chr18.fastq.gz
Approx 80% complete for SRR3099587_chr18.fastq.gz
Approx 85% complete for SRR3099587_chr18.fastq.gz
Approx 90% complete for SRR3099587_chr18.fastq.gz
Approx 95% complete for SRR3099587_chr18.fastq.gz
Analysis complete for SRR3099587_chr18.fastq.gz
[Tue Feb 20 14:22:57 2024]
Finished job 1.
1 of 3 steps (33%) done
Select jobs to execute...
Execute 1 jobs...
[Tue Feb 20 14:22:57 2024]
localrule multiqc:
input: FastQC/SRR3099585_chr18_fastqc.zip, FastQC/SRR3099586_chr18_fastqc.zip, FastQC/SRR3099587_chr18_fastqc.zip
output: multiqc_report.html, multiqc_data
jobid: 2
reason: Input files updated by another job: FastQC/SRR3099586_chr18_fastqc.zip, FastQC/SRR3099585_chr18_fastqc.zip, FastQC/SRR3099587_chr18_fastqc.zip
resources: tmpdir=/var/tmp/pbs.743371.pbsserver
multiqc FastQC/SRR3099585_chr18_fastqc.zip FastQC/SRR3099586_chr18_fastqc.zip FastQC/SRR3099587_chr18_fastqc.zip
[WARNING] multiqc : MultiQC Version v1.20 now available!
[INFO ] multiqc : This is MultiQC v1.9
[INFO ] multiqc : Template : default
[INFO ] multiqc : Searching : /data/work/I2BC/chloe.quignot/snakemake_tutorial/FastQC/SRR3099585_chr18_fastqc.zip
[INFO ] multiqc : Searching : /data/work/I2BC/chloe.quignot/snakemake_tutorial/FastQC/SRR3099586_chr18_fastqc.zip
[INFO ] multiqc : Searching : /data/work/I2BC/chloe.quignot/snakemake_tutorial/FastQC/SRR3099587_chr18_fastqc.zip
[INFO ] fastqc : Found 3 reports
[INFO ] multiqc : Compressing plot data
[INFO ] multiqc : Report : multiqc_report.html
[INFO ] multiqc : Data : multiqc_data
[INFO ] multiqc : MultiQC complete
[Tue Feb 20 14:23:02 2024]
Finished job 2.
2 of 3 steps (67%) done
Select jobs to execute...
Execute 1 jobs...
[Tue Feb 20 14:23:02 2024]
localrule all:
input: FastQC/SRR3099585_chr18_fastqc.html, FastQC/SRR3099586_chr18_fastqc.html, FastQC/SRR3099587_chr18_fastqc.html, FastQC/SRR3099585_chr18_fastqc.zip, FastQC/SRR3099586_chr18_fastqc.zip, FastQC/SRR3
099587_chr18_fastqc.zip, multiqc_report.html, multiqc_data
jobid: 0
reason: Input files updated by another job: multiqc_data, multiqc_report.html, FastQC/SRR3099585_chr18_fastqc.html, FastQC/SRR3099586_chr18_fastqc.zip, FastQC/SRR3099585_chr18_fastqc.zip, FastQC/SRR309
9587_chr18_fastqc.html, FastQC/SRR3099587_chr18_fastqc.zip, FastQC/SRR3099586_chr18_fastqc.html
resources: tmpdir=/var/tmp/pbs.743371.pbsserver
[Tue Feb 20 14:23:02 2024]
Finished job 0.
3 of 3 steps (100%) done
Complete log: .snakemake/log/2024-02-20T142240.002684.snakemake.log
Observe the output
Snakemake detects that the output files of SRR3099587_chr18 are missing (“reason: Missing output files: FastQC/SRR3099587_chr18_fastqc.html, FastQC/SRR3099587_chr18_fastqc.zip“) and also that “Set of input files has changed since last execution” so it re-ran the fastqc rule.
Have a look at your output folder, you should now have 6 files in there:
john.doe@node06:/data/work/I2BC/john.doe/snakemake_tutorial$ ls FastQC
SRR3099585_chr18_fastqc.html SRR3099586_chr18_fastqc.html SRR3099587_chr18_fastqc.html
SRR3099585_chr18_fastqc.zip SRR3099586_chr18_fastqc.zip SRR3099587_chr18_fastqc.zip
Have you noticed that, up until now, Snakemake re-executed FastQC on all inputs every time the fastqc rule was run, indifferent of the fact that the output files were already generated or not?
No? Have a closer look at the output log that you just got… In fact, if you look closely, fastqc is always run only once but on a variable number of inputs (depending on the input of our rule):
Job stats:
job count
------- -------
all 1
fastqc 1
multiqc 1
total 3
...
fastqc --outdir FastQC Data/SRR3099585_chr18.fastq.gz Data/SRR3099586_chr18.fastq.gz Data/SRR3099587_chr18.fastq.gz
Snakemake re-runs fastqc on all inputs because we gave as input to the fastqc rule a list of files rather than just a single file. However, this is sub-optimal, especially on a computer cluster on which you have access to a large amount of available resources to run your jobs on. We’ll see how to change this in the next objective.