Example files are accessible on Zenodo as a tar.gz archive ![]() . To fetch and extract the files, you can use the following command lines:
. To fetch and extract the files, you can use the following command lines:
We will be working on the I2BC cluster, on which all the tools that we need are already installed. To connect to the cluster you will need your Multipass login information. Please refer to Step 4 on the I2BC cluster training page for more details on how to proceed.
Once on the cluster, connect to a node (that’s where all the tools are installed):
john.doe@cluster-i2bc:~$ qsub -I -q common
qsub: waiting for job 659602.pbsserver to start
qsub: job 659602.pbsserver ready
john.doe@node06:~$
Then load the appropriate tools using the module command:
module load snakemake/snakemake-8.4.6
module load fastqc/fastqc_v0.11.5
module load nodes/multiqc-1.9
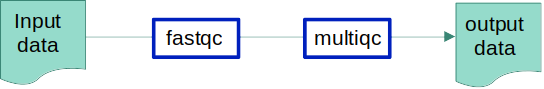
Why these 3 tools? Because we will be using all three in our pipeline.
Double-check that you’ve loaded the modules correctly, for example, by checking their version:
john.doe@node06:~$ snakemake --version
8.4.6
And create & move to your chosen working space, for example:
WORKDIR="/data/work/I2BC/$USER/snakemake_tutorial"
mkdir -p $WORKDIR
cd $WORKDIR
Example files are accessible on Zenodo as a tar.gz archive ![]() . To fetch and extract the files, you can use the following command lines:
. To fetch and extract the files, you can use the following command lines:
# download archive
wget "https://zenodo.org/record/3997237/files/FAIR_Bioinfo_data.tar.gz"
# extract files
tar -zxf FAIR_Bioinfo_data.tar.gz
# delete the archive
rm FAIR_Bioinfo_data.tar.gz
You should now see in your working space, a folder called Data containing compressed fastq files (*.fastq.gz) and O. tauri genome files (*.gff and *.fna):
john.doe@node06:/data/work/I2BC/john.doe/snakemake_tutorial$ ls Data/
O.tauri_annotation.gff SRR3099585_chr18.fastq.gz SRR3099587_chr18.fastq.gz SRR3105698_chr18.fastq.gz
O.tauri_genome.fna SRR3099586_chr18.fastq.gz SRR3105697_chr18.fastq.gz SRR3105699_chr18.fastq.gz