Getting started with Snakemake
Let’s understand the bash script & identify all the steps and tools involved that we will need
Let’s split the bash script into parts:
The first part is applied once, at the beginning of the script in order to index the reference genome that will be used to map the reads:
# line 30
bowtie2-build --threads ${ncpus_build} ${genome} Bwt2_index/tauri > Bwt2_index/Bwt2_index.log 2>&1
# input: reference genome fasta
# tool: bowtie2-build (bowtie/bowtie2-2.2.9)
# aim: index the reference genome
# output: Bwt2_index/ folder with indexed genome files
In the “for” loop section, we apply on each sample independently a set of tools:
# line 43
fastqc --outdir FastQC ${rnadir}/${sample}.fastq.gz > FastQC/${sample}.log 2>&1
# input: each RNAseq fastq
# tool: fastqc (fastqc/fastqc_v0.11.5)
# aim: run QC analysis (per sample)
# output: a zip & html report in FastQC/ folder
# line 47
bowtie2 --threads ${ncpus_map} -x Bwt2_index/tauri -U ${rnadir}/${sample}.fastq.gz -S SAMs/${sample}.sam > LOGs/${sample}_bowtie2.log 2>&1
# input: each RNAseq fastq + the indexed reference genome files
# tool: bowtie2 (bowtie/bowtie2-2.2.9)
# aim: map the reads onto the reference genome (per sample)
# output: mapped reads in SAM format within the SAMs/ folder
# lines 51-53
samtools view -b SAMs/${sample}.sam -o BAMs/${sample}.bam
samtools sort --threads ${ncpus_sort} BAMs/${sample}.bam -o BAMs/${sample}_sort.bam
samtools index -@ ${ncpus_index} BAMs/${sample}_sort.bam
# input: mapped reads in SAM format
# tool: samtools (nodes/samtools-1.11)
# aim: 1. change format sam -> bam (per sample)
# 2. sort reads (per sample)
# 3. index the sorted bam (per sample)
# general output: sorted and indexed reads in BAM format within the BAMs/ folder
# line 57
featureCounts -T ${ncpus_count} -t gene -g ID -a ${annots} -s 2 -o Counts/${sample}_ftc.txt BAMs/${sample}_sort.bam > LOGs/${sample}_ftc.log 2>&1
# input: sorted & indexed reads in BAM format + reference genome annotation
# tool: featureCounts (subread/subread-1.5.2)
# aim: count reads per gene in reference genome (per sample)
# output: a gene count table within the Counts/ folder
Outside the for loop, at the end of the bash pipeline, we concatenate all the count tables:
# lines 61-62
paste Counts/*_ftc.txt > Counts/ftc_tmp.txt
awk -v nb=${nbs} -v col=7 'BEGIN {FS ="\t"}{ctmp=$1; for (i=col; i<=nb*col; i=i+col) {count=sprintf("%s\t%s", ctmp, $i); ctmp=count}; print count}' Counts/ftc_tmp.txt | sed '1d' > Counts/counts.txt
# input: gene count table of all samples
# tools: paste & awk (standard in bash)
# aim: concatenate the gene count table of all samples and reformat to a clean tab-separated file
# output: a single clean tab-separated gene count table containing all samples
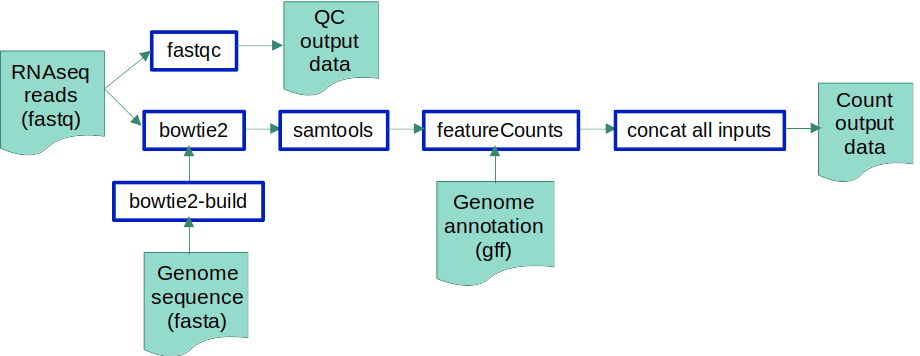
If we visualise the pipeline with the tools, we should have something like this:
If you try running the bash script, make sure you to load the modules first:
module load fastqc/fastqc_v0.11.5 subread/subread-1.5.2 bowtie/bowtie2-2.2.9 nodes/samtools-1.11
NB: subread is the package that contains featureCounts
john.doe@node06:~$ featureCounts -v
featureCounts v1.5.2
john.doe@node06:~$ fastqc --version
FastQC v0.11.5
john.doe@node06:~$ samtools --version
samtools 1.11
Using htslib 1.11-4
Copyright (C) 2020 Genome Research Ltd.
john.doe@node06:~$ bowtie2 --version
/usr/bin/bowtie2-align-s version 2.4.2
64-bit
Execution example (copy the script to your directory first, here we named it
bash_pipeline.sh):
bash bash_pipeline.sh -g Data/O.tauri_genome.fna -a Data/O.tauri_annotation.gff -d Data/ SRR3099585_chr18 SRR3099586_chr18 SRR3099587_chr18 SRR3105697_chr18 SRR3105698_chr18 SRR3105699_chr18
See the IFB’s training session pdf for step by step help: