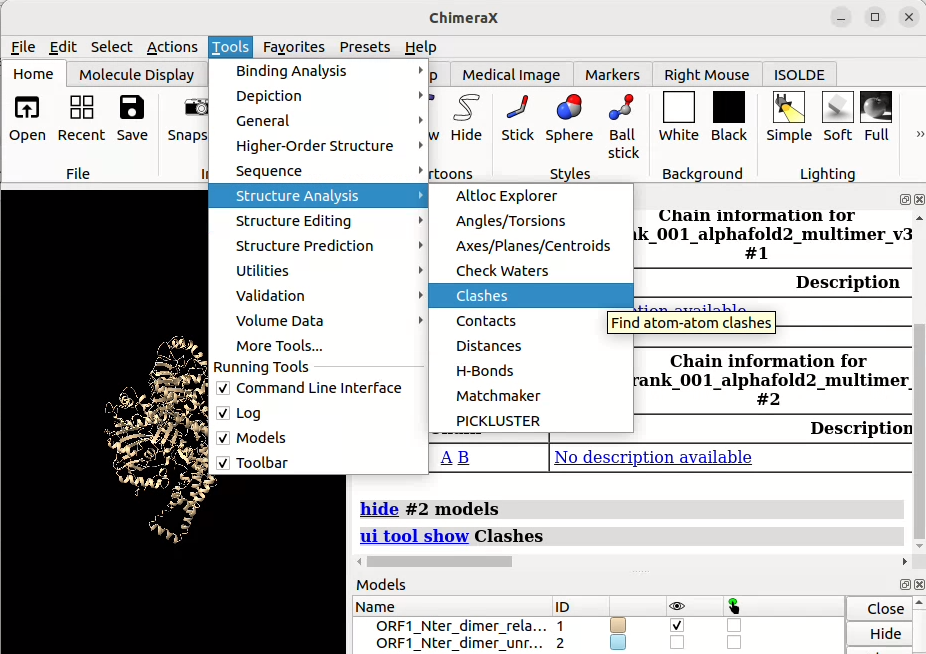
![]() graphically through the Menu bar in (/!\ make sure you only have one model active)
graphically through the Menu bar in (/!\ make sure you only have one model active)
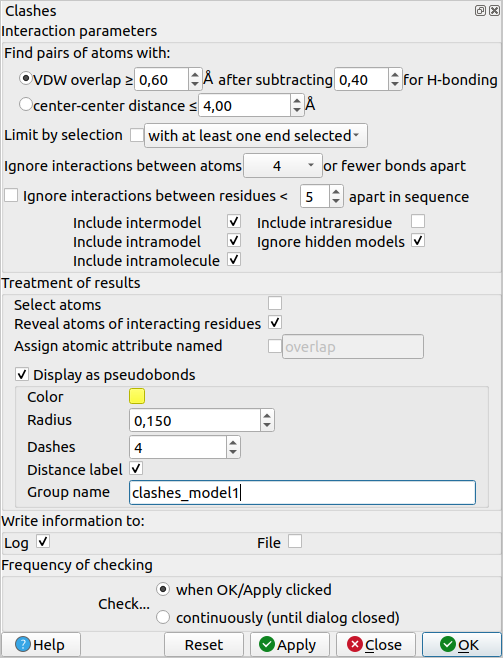
![]() in-line with the clashes command, for example:
in-line with the clashes command, for example:
![]() Click here for detailed information on the clashes command line options.
Click here for detailed information on the clashes command line options.
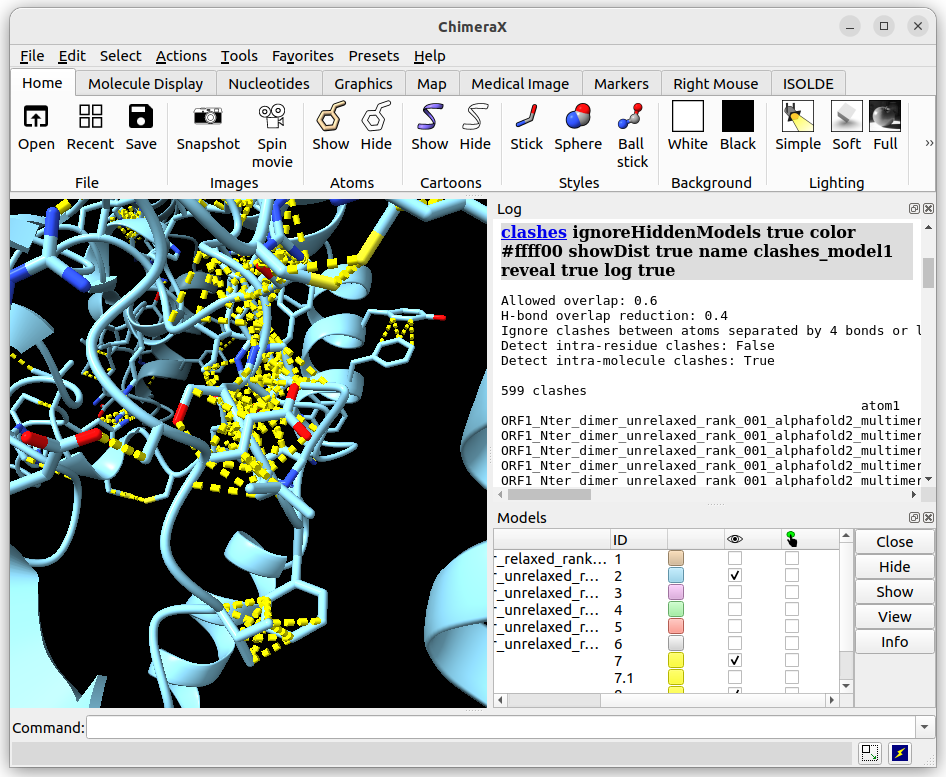
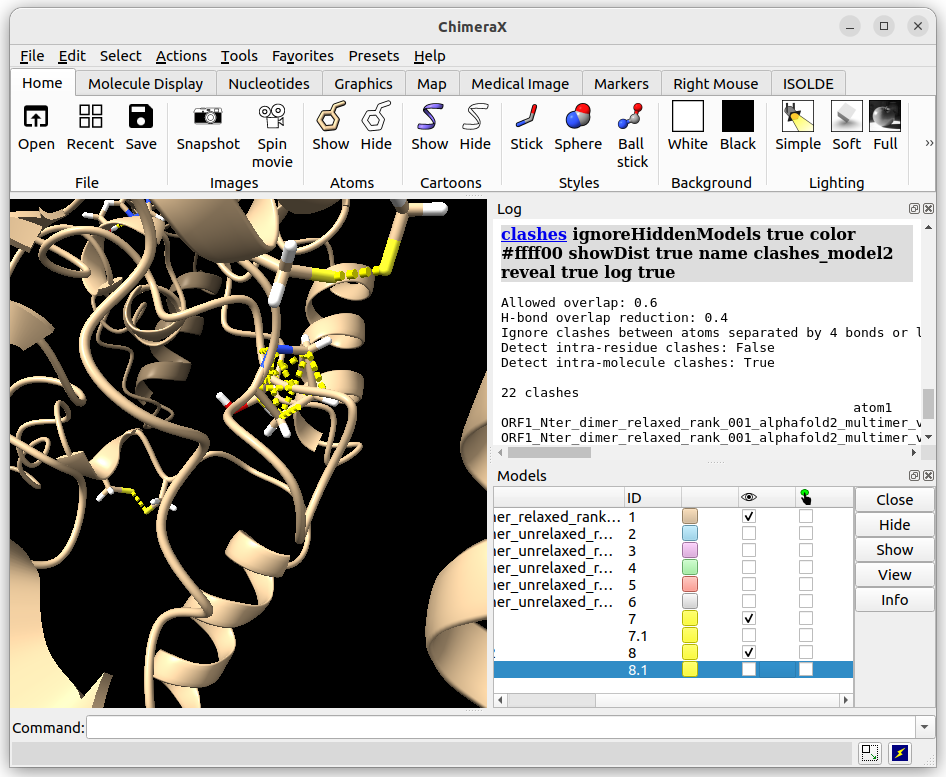
![]() graphically through the Menu bar in
graphically through the Menu bar in
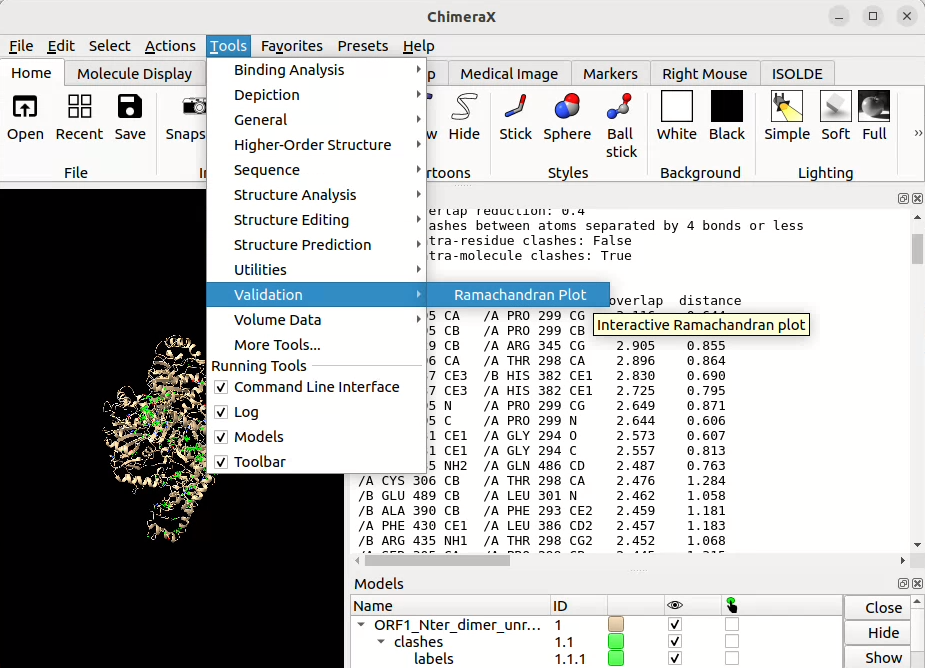
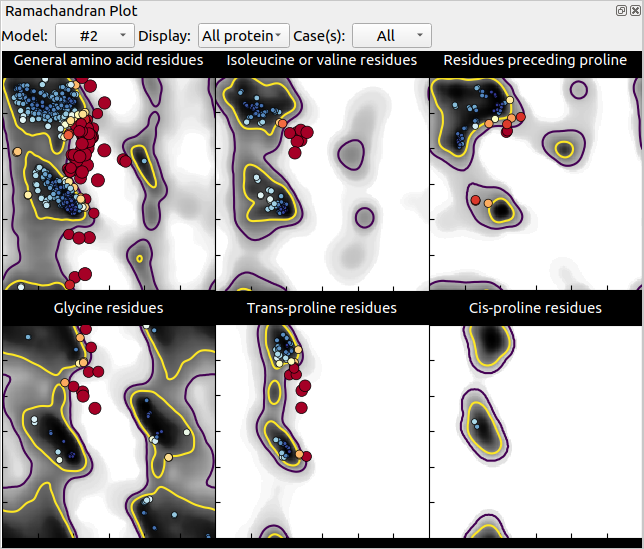
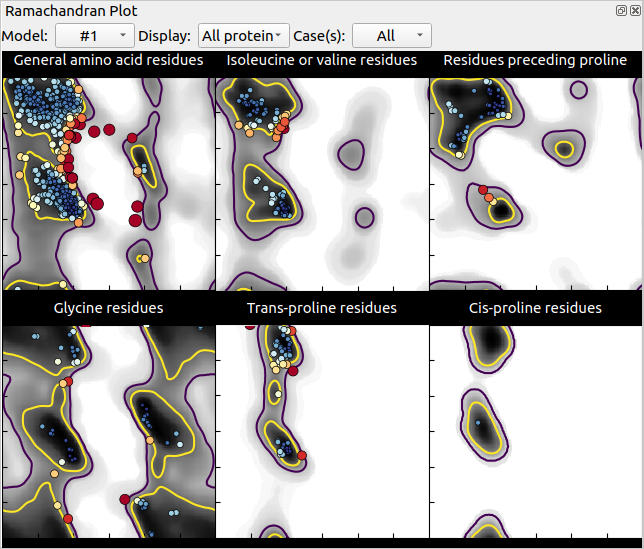
![]() plotting Ramachandran maps through the command line doesn’t seem to be possible (yet) with ISOLDE…
plotting Ramachandran maps through the command line doesn’t seem to be possible (yet) with ISOLDE…
![]() Click here for detailed information on the Ramachandran plot in ISOLDE.
Click here for detailed information on the Ramachandran plot in ISOLDE.