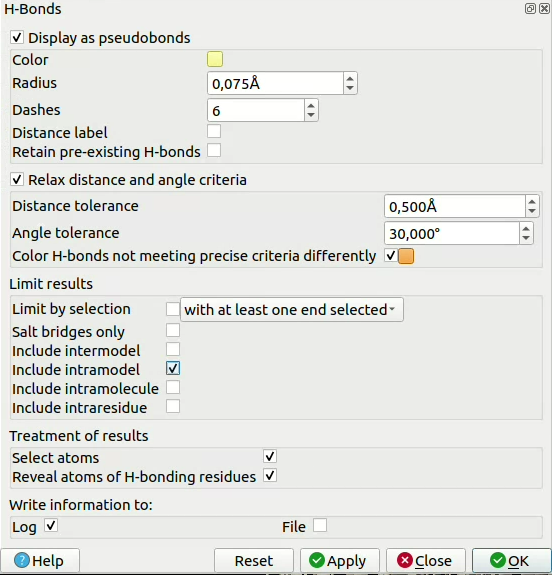
![]() click on , then select the parameters you want. You can, for example, relax the constraints on angles and distances to allow for imperfect models. You can also define here if you want to restrict yourself to bonds between two different chains (“Include intramodel” ticked and the rest unticked) or if you would like to include intermolecule bonds too.
click on , then select the parameters you want. You can, for example, relax the constraints on angles and distances to allow for imperfect models. You can also define here if you want to restrict yourself to bonds between two different chains (“Include intramodel” ticked and the rest unticked) or if you would like to include intermolecule bonds too.
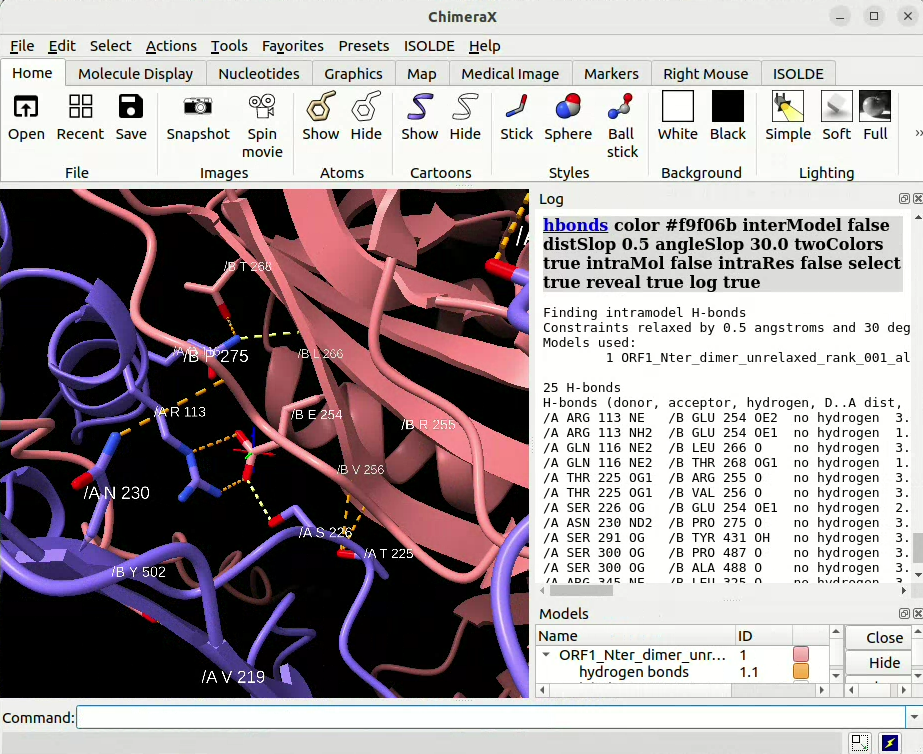
![]() in-line you can use the hbonds command to calculate H-bonds and/or Salt bridges, for example:
in-line you can use the hbonds command to calculate H-bonds and/or Salt bridges, for example:
![]() Click here for more information on the hbonds command line options.
Click here for more information on the hbonds command line options.
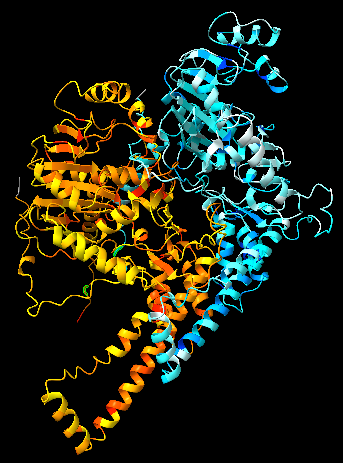
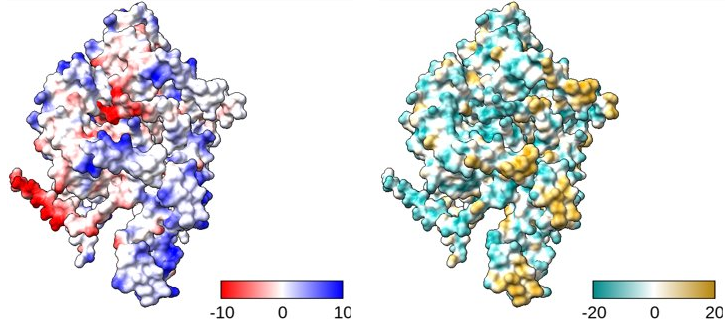
![]() click on the tab in the Menu bar, then select “electrostatic” or “hydrophobic” in the section to colour your model by electrostatic charges or hydrophobicity.
click on the tab in the Menu bar, then select “electrostatic” or “hydrophobic” in the section to colour your model by electrostatic charges or hydrophobicity.
![]() in-line you can use the coulombic and mlp commands to colour by electrostatics and hydrophobicity respectively, for example:
in-line you can use the coulombic and mlp commands to colour by electrostatics and hydrophobicity respectively, for example:
![]() click on the tab in the Menu bar, then select “Open” and upload the multiple sequence alignment (MSA) file outputted by AlphaFold in fasta format. This will open an additionnal pane with your MSA. Right click on it, go in to select the models and chains the MSA covers.
click on the tab in the Menu bar, then select “Open” and upload the multiple sequence alignment (MSA) file outputted by AlphaFold in fasta format. This will open an additionnal pane with your MSA. Right click on it, go in to select the models and chains the MSA covers.